Publications
2025
-
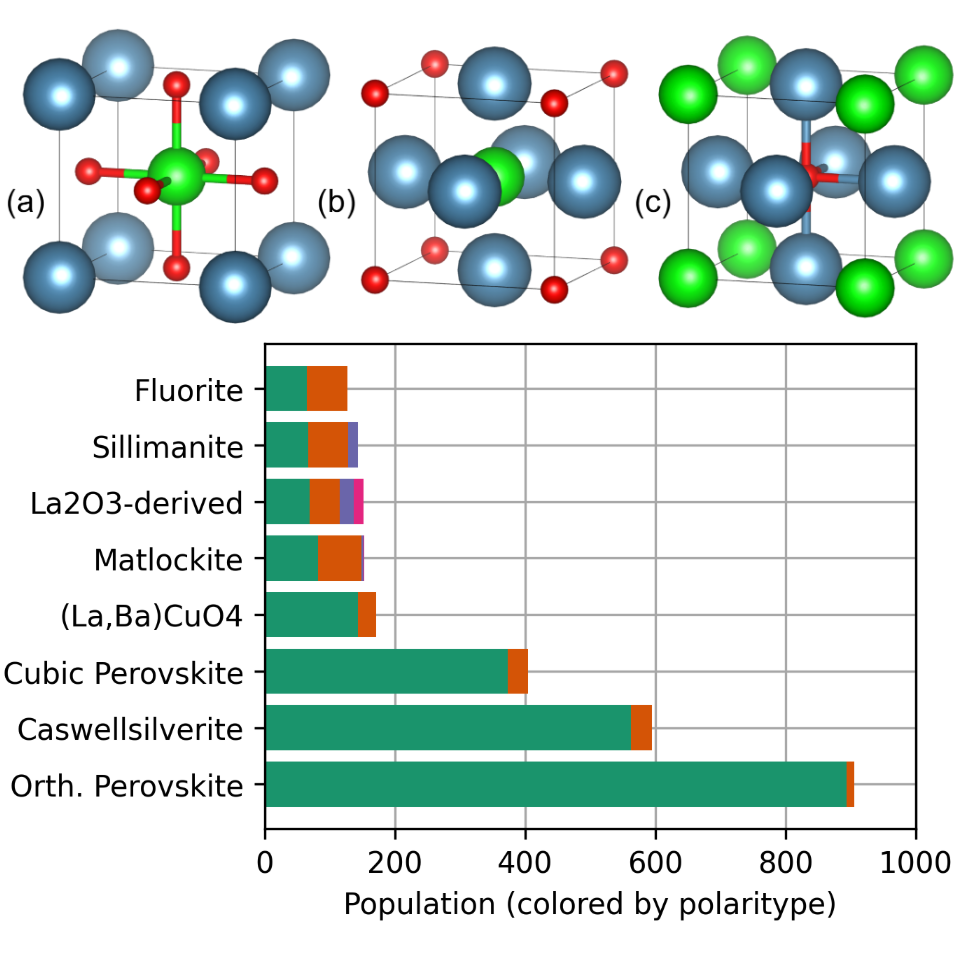 Decoratypes: An Extensible Crystal Taxonomy for Machine Learning-Guided Materials DiscoveryKyle D. Miller , Michele Campbell , Danilo Puggioni , and James M. RondinelliPhysical Review Materials, Oct 2025
Decoratypes: An Extensible Crystal Taxonomy for Machine Learning-Guided Materials DiscoveryKyle D. Miller , Michele Campbell , Danilo Puggioni , and James M. RondinelliPhysical Review Materials, Oct 2025We introduce decoratypes, a structure taxonomy that classifies compounds based on site decorations of specific structural prototypes. Building on this foundation, a ferroelectric materials discovery framework is developed, integrating decoratypes with an active learning approach to accelerate exploration. In addition, six novel ferroelectric candidates are predicted, including three strain-activated ferroelectrics and three strain-activated hyperferroelectrics. These findings highlight the potential of the decoratype taxonomy to enhance our understanding of structure-driven material properties and facilitate the discovery of promising yet underexplored regions of chemical space.
-
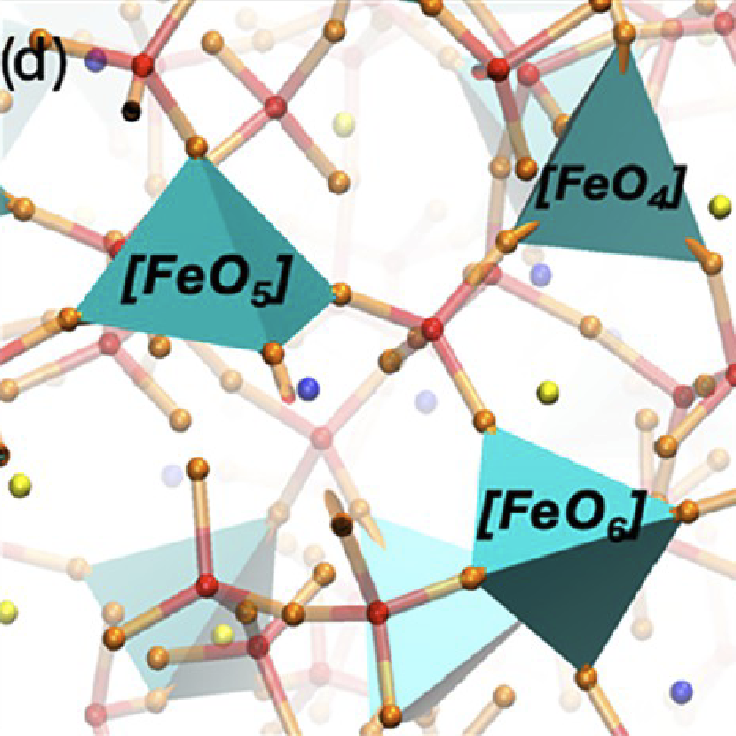 Revealing the Structure–Property Relationships of Nd\(_2\)O\(_3\) -Containing Alkali Iron Phosphate Glasses from Molecular Dynamics SimulationsJayani Kalahe , Xiaonan Lu , Kyle Miller , Miroslava Peterson , Brian J. Riley , John D. Vienna , James E. Saal , and Jincheng DuChemistry of Materials, Oct 2025
Revealing the Structure–Property Relationships of Nd\(_2\)O\(_3\) -Containing Alkali Iron Phosphate Glasses from Molecular Dynamics SimulationsJayani Kalahe , Xiaonan Lu , Kyle Miller , Miroslava Peterson , Brian J. Riley , John D. Vienna , James E. Saal , and Jincheng DuChemistry of Materials, Oct 2025The atomic structure and structure-property relationships of Nd2O3-containing potassium iron phosphate glasses were investigated by using molecular dynamics (MD) simulations based on nulcear waste glass compositions designed with artificial intellegence (AI) and machine learning (ML) approaches. The results show that Nd3+ ions act as modifiers, with an average Nd3+-O bond distance of 2.35 Å and a coordination number of approximately 6. Nd3+ ions were found to be preferentially associated with phosphate anionic units such as [PO4]3- and [P2O7]4-, leading to the formation of nonbridging oxygens, while K+ ions primarily compensate for the negatively charged [FeO4]- units. Clustering of Nd3+ ions was observed in the simulated glasses, with a decrease in homogeneous dispersion as the Nd2O3 concentration increased from 5 to 10 mol %. Additionally, increasing the P2O5 content increases the fraction of isolated Nd3+ ions. The addition of Nd2O3 notably affected the glass structures and led to significant changes in the mechanical properties and glass transition temperature values. The results demonstrate that physics based modeling method such as MD simulations with reliable interatomic potentials can be a valuable tool for composition optimization based on AI/ML design and for elucidating the complex structures and structure- property relationships of rare-earth-ion-containing phosphate glasses, a materials class with applications in critical fields such as photonics and nuclear waste disposal.
-
 Glass-Bonded Monazite Waste Forms for Lanthanide and Actinide Immobilization: From Theoretical Design to Scale-Up Production and CharacterizationMiroslava Peterson , Brian J. Riley , Xiaonan Lu , Kayla H. Yano , Semanti Mukhopadhyay , Ian I. Leavy , Ethan K. Nickerson , Robert J. Seffens , Jincheng Du , Kyle D. Miller , and James E. SaalACS Omega, Aug 2025
Glass-Bonded Monazite Waste Forms for Lanthanide and Actinide Immobilization: From Theoretical Design to Scale-Up Production and CharacterizationMiroslava Peterson , Brian J. Riley , Xiaonan Lu , Kayla H. Yano , Semanti Mukhopadhyay , Ian I. Leavy , Ethan K. Nickerson , Robert J. Seffens , Jincheng Du , Kyle D. Miller , and James E. SaalACS Omega, Aug 2025The development of nuclear waste forms for both existing and future nuclear wastes is critical to ensuring global environmental safety. This study focuses on waste management from molten salt reactors, where fuel exists in a salt form and could be processed in real time for the removal of neutron poisons such as xenon isotopes (e.g., 135Xe) and rare earth elements (REEs, e.g., 149Sm). To ensure safe, stable, and long-term disposal in geological repositories, REEs must be incorporated into a durable waste form. Iron-phosphate glasses are a promising candidate due to their low melting points, high chemical durability, and their ability to incorporate high concentrations of REEs. In this study, we successfully prepared iron-phosphate glass waste forms with high Nd loadings (up to 37 mass %) in batch sizes ranging from small (23 g) to large (1600 g). The resulting materials contained up to 75 mass % NdPO4, contributing to their mechanical resilience and exceptional chemical durability. These findings highlight the potential of iron-phosphate glasses as high-efficiency, chemically durable waste forms and demonstrate the successful transition from theoretical design to scaled-up production.



2024
-
 Design and Discovery of Metal-Insulator Transition MaterialsKyle Daniel MillerNorthwestern University , Aug 2024
Design and Discovery of Metal-Insulator Transition MaterialsKyle Daniel MillerNorthwestern University , Aug 2024Metal-insulator transitions (MITs) are a resistive switching phenomenon arising from the interplay of lattice, electronic, and magnetic forces. This switching functionality has the potential to revolutionize device architecture for applications such as memory, communication, photodetection, and neuromorphic computing. Decades of research have produced a wealth of insight into the coupling between the driving forces, enabling functionalization of these transitions to suit various device applications. The most notable example, VO\(_{2}\), exhibits an MIT near room temperature which is tunable via chemical doping, photodoping, strain engineering, thickness control, and applied electric field. Despite these successes, many of the transitions are still not fully understood and the pool of known MIT compounds remains small.\ To address these remaining challenges, I employ simulation and informatics methods to elucidate structure-property relationships and develop strategies for designing and discovering MIT compounds. Motivated by the existence of two known MIT materials, VO\(_{2}\) and NbO\(_{2}\), in the rutile family, I start by investigating the trirutile structure, a rutile superstructure providing additional opportunities for tuning. Leveraging first-principles calculations, I explore MgTa\(_{2}\)O\(_{6}\) as a model system for extending functionalization of Peierls MIT behavior from rutile oxides to trirutile oxides. Near a d\(^{1}\) electron concentration, I identify an insulating state enabled by metal-metal dimerization and characterized by a 0.625 axial ratio, both key features of the insulating state found in natively d\(^{1}\) rutile oxide MIT materials. Symmetry analysis reveals that the trirutile structure can accommodate this Peierls-like transition without the symmetry breaking required in the rutile structure. These findings highlight the trirutile superstructure as a new frontier for engineering Peierls MITs.\ In another MIT compound, the Ni-doped BaCo\(_{1-x}\)Ni\(_x\)S\(_2\) system, our experimental collaborators observed anomalous atomic displacements arising near the MIT phase boundary. Using first-principles calculation, I build a theoretical model that explains the origin of these displacements. I identify a previously unreported first-order Jahn-Teller instability which connects the atomic displacements to orbital symmetry-breaking that promotes the insulating state. Both effects are suppressed to allow metallization upon electron doping via Ni substitution, highlighting the role of structure in an MIT previously described as purely electronic. Furthermore, I derive competing orbital-structural orderings to provide an explanation for the anisotropy and disorder identified by our collaborator’s in-situ observations. This case study of BaCo\(_{1-x}\)Ni\(_x\)S\(_2\) shows the importance of considering structural contributions to MITs even when strong electronic driving forces are present.\ Finally, I evaluate the utility and limitations of the global instability index (GII), a low-cost bond strain metric found to be a useful descriptor in prediction of MIT behavior. Analyzing GII values for >32,000 compounds, I find some correlation with stability, but also sensitivity to chemistry, geometry, data source, and the underlying bond valence model, complicating its use as a universal stability indicator. From these insights, I offer recommendations for more robust usage and suggest how the metric can help us identify MIT-susceptible compounds.\ Overall, this work demonstrates the power of combining data-driven and physics-based approaches to describe and model the complexities of correlated electron systems like MITs. We demonstrate the use of both top-down and bottom-up approaches drawing from in-situ and in-silico data to develop materials design tools and strategies.

2023
-
 Machine Learning the Electronic Structure of Matter across TemperaturesLenz Fiedler , Normand A. Modine , Kyle D. Miller , and Attila CangiPhysical Review B, Sep 2023
Machine Learning the Electronic Structure of Matter across TemperaturesLenz Fiedler , Normand A. Modine , Kyle D. Miller , and Attila CangiPhysical Review B, Sep 2023We introduce machine learning (ML) models that predict the electronic structure of materials across a wide temperature range. Our models employ neural networks and are trained on density functional theory (DFT) data. Unlike most other ML models that use DFT data, our models directly predict the local density of states (LDOS) of the electronic structure. This provides several advantages, including access to multiple observables such as the electronic density and electronic total free energy. Moreover, our models account for both the electronic and ionic temperatures independently, making them ideal for applications like laser heating of matter. We validate the efficacy of our LDOS-based models on a metallic test system. They accurately capture energetic effects induced by variations in ionic and electronic temperatures over a broad temperature range, even when trained on a subset of these temperatures. These findings open up exciting opportunities for investigating the electronic structure of materials under both ambient and extreme conditions.
-
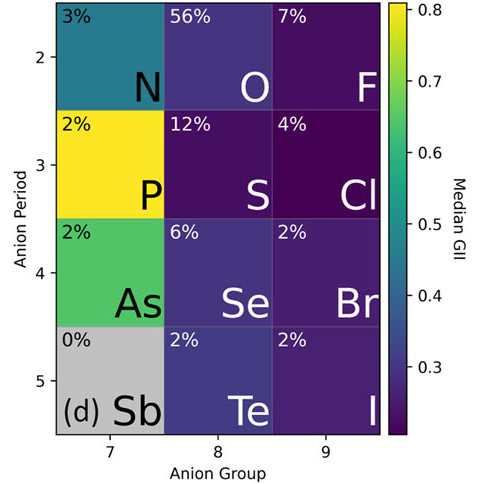 Testing the Limits of the Global Instability IndexKyle D. Miller , and James M. RondinelliAPL Materials, Oct 2023
Testing the Limits of the Global Instability IndexKyle D. Miller , and James M. RondinelliAPL Materials, Oct 2023The global instability index (GII) is a computationally inexpensive bond valence-based metric originally designed to evaluate the total bond strain in a crystal. Recently, the GII has gained popularity as a feature of data-driven models in materials research. Although prior studies have proven that GII is an effective predictor of structural distortions and decomposition energy when applied to small datasets, the wider use of GII as a global indicator of structural stability has yet to be evaluated. To that end, we compute GII for thousands of compounds in inorganic structure databases and partition compounds by chemical interactions underlying their stability to understand the GII values and their variations. Our results show that the GII captures relative chemical trends, such as electronegativity, even beyond the intended domain of strongly ionic compounds. However, we also find that GII magnitudes vary significantly with factors such as chemistry (cation–anion identities and bond character), geometry (connectivity), data source, and model bias, making GII suitable for comparisons within controlled datasets but unsuitable as an absolute, universal metric for structural feasibility.


2022
-
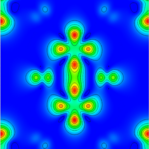 Carrier-Induced Metal-Insulator Transition in Trirutile MgTa\(_2\)O\(_6\)Kyle D. Miller , and James M. RondinelliPhysical Review Materials, Jul 2022
Carrier-Induced Metal-Insulator Transition in Trirutile MgTa\(_2\)O\(_6\)Kyle D. Miller , and James M. RondinelliPhysical Review Materials, Jul 2022Materials exhibiting metal-insulator transitions (MITs) are proposed platforms for next-generation low-power electronics. Many of these materials exhibit strong coupling between the electronic and lattice degrees of freedom, which makes them ideal systems to examine the interplay between lattice dynamics, electronic structure, and magnetic order. The rutile structure, featuring edge-connected octahedral chains along its c axis, permits metal-metal interactions that can induce metal-insulator transitions. Although the MITs in rutile-structured compounds have been thoroughly studied, the derivative trirutile phase, which accommodates similar metal-metal interactions, has yet to be examined in the context of MITs. Here we use density functional theory calculations to investigate a carrier-driven MIT in trirutile MgTa\(_2\)O\(_6\), a \(d^0\) insulator with a suitable axial ratio. Our calculations suggest the existence of four distinct phases in MgTa\(_2\)O\(_6\) with increasing electron concentration: nonmagnetic insulator, ferromagnetic (FM) metal, FM half-metal, and nonmagnetic insulator. We explain how the resulting electronic phases arise from changes in atomic structure with increasing carrier density. Our results indicate that trirutile oxides may be a promising materials class for which to access and functionalize MITs.

2021
-
 Database, Features, and Machine Learning Model to Identify Thermally Driven Metal–Insulator Transition CompoundsAlexandru B. Georgescu , Peiwen Ren , Aubrey R. Toland , Shengtong Zhang , Kyle D. Miller , Daniel W. Apley , Elsa A. Olivetti , Nicholas Wagner , and James M. RondinelliChemistry of Materials, Jul 2021
Database, Features, and Machine Learning Model to Identify Thermally Driven Metal–Insulator Transition CompoundsAlexandru B. Georgescu , Peiwen Ren , Aubrey R. Toland , Shengtong Zhang , Kyle D. Miller , Daniel W. Apley , Elsa A. Olivetti , Nicholas Wagner , and James M. RondinelliChemistry of Materials, Jul 2021Metal–insulator transition (MIT) compounds are materials that may exhibit metallic or insulating behavior, depending on the physical conditions, and are of immense fundamental interest owing to their potential applications in emerging microelectronics. An important subset of MIT materials are those with a transition driven by temperature. The number of thermally driven MIT materials, however, is scarce, which makes delineating these compounds from those that are exclusively insulating or metallic challenging. Most research that addresses thermal MITs is limited by the domain knowledge of the scientists to a subset of MIT materials and is often focused on a limited subset of possible features. Here, using a combination of domain knowledge and natural language processing (NLP) searches, we have built a material database comprising thermally driven MITs as well as metals and insulators with similar chemical composition and stoichiometries to the MIT compounds. We featurized this data set using a wide variety of compositional, structural, and energetic descriptors, including two MIT relevant energy scales, the estimated Hubbard interaction and the charge transfer energy, as well as the structure-bond-stress metric referred to as the global-instability index (GII). We then performed supervised classification on this data set, constructing three electronic-state classifiers: metal vs nonmetal (M), insulator vs noninsulator (I), and MIT vs non-MIT (T). This classification allows us to identify new features separating MIT materials from non-MIT materials. These include the 2D feature space consisting of the average deviation of the covalent radius, the range of the Mendeleev number and Ewald energy. We discuss the relationship of these atomic features to the physical interactions underlying MITs in the rare-earth nickelate family. We then elaborate on other features (GII and Ewald energy) and examine how they affect the classification of binary vanadium and titanium oxides. Last, we implement an online and publicly accessible version of the classifiers, enabling quick probabilistic class predictions by uploading a crystallographic structure file. The broad accessibility of our database, newly identified features, and user-friendly classifier models will aid in accelerating the discovery of MIT materials.
-
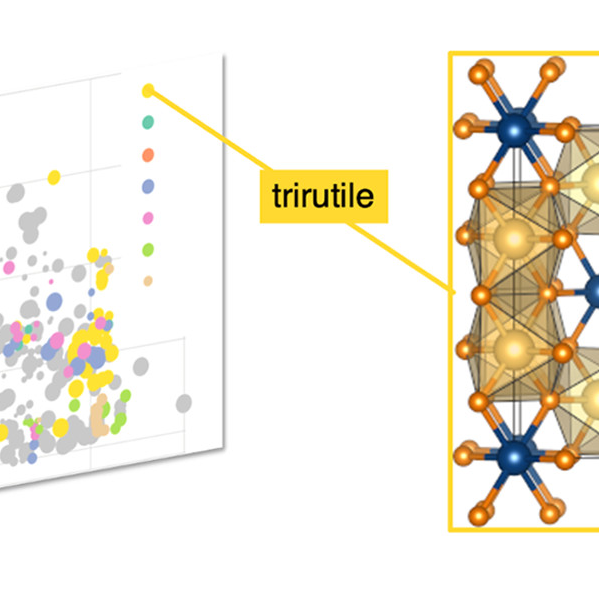 AB\(_2\)X\(_6\) Compounds and the Stabilization of Trirutile OxidesEmily C. Schueller , Yuzki M. Oey , Kyle D. Miller , Kira E. Wyckoff , Ruining Zhang , William Zhang , Stephen D. Wilson , James M. Rondinelli , and Ram SeshadriInorganic Chemistry, Jun 2021
AB\(_2\)X\(_6\) Compounds and the Stabilization of Trirutile OxidesEmily C. Schueller , Yuzki M. Oey , Kyle D. Miller , Kira E. Wyckoff , Ruining Zhang , William Zhang , Stephen D. Wilson , James M. Rondinelli , and Ram SeshadriInorganic Chemistry, Jun 2021The properties of crystalline materials tend to be strongly correlated with their structures, and the prediction of crystal structure from only the composition is a coveted goal in the field of inorganic materials. However, even for the simplest compositions, such prediction relies on a complex network of interactions, including atomic or ionic radii, ionicity, electronegativity, position in the periodic table, and magnetism, to name only a few important parameters. We focus here on the AB\(_2\)X\(_6\) (AB\(_2\)O\(_6\) and AB\(_2\)F\(_6\)) composition space with the specific goal of finding new oxide compounds in the trirutile family, which is known for unusual one-dimensional (1D) antiferromagnetic behavior. Through machine learning methods, we develop an understanding of how geometric and bonding constraints determine the crystallization of compounds in the trirutile structure as opposed to other ternary structures in this space. In combination with density functional theory (DFT) calculations, we predict 16 previously unreported candidate trirutile oxides. We successfully prepare one of these and show it forms in the disordered rutile structure, under the preparation conditions adopted here.


2020
-
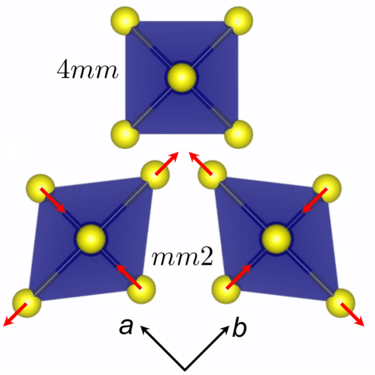 Structural Signatures of the Insulator-to-Metal Transition in BaCo\(_{1-x}\)Ni\(_x\)S\(_2\)Emily C. Schueller , Kyle D. Miller , William Zhang , Julia L. Zuo , James M. Rondinelli , Stephen D. Wilson , and Ram SeshadriPhysical Review Materials, Oct 2020
Structural Signatures of the Insulator-to-Metal Transition in BaCo\(_{1-x}\)Ni\(_x\)S\(_2\)Emily C. Schueller , Kyle D. Miller , William Zhang , Julia L. Zuo , James M. Rondinelli , Stephen D. Wilson , and Ram SeshadriPhysical Review Materials, Oct 2020The solid solution BaCo\(_{1-x}\)Ni\(_x\)S\(_2\) exhibits an insulator-to-metal transition close to \(x=0.21\). Questions of whether this transition is coupled with structural changes remain open. Here we follow the structural evolution as a function of the Ni content \(x\) using synchrotron powder x-ray diffraction and pair distribution function analyses to reveal significant basal sulfide anion displacements occurring preferentially along the CoS\(_5\) pyramidal edges comprising the edge-connected bond network in BaCo\(_{1-x}\)Ni\(_x\)S\(_2\). These displacements decrease in magnitude as \(x\) increases and are nearly quenched in \(x=1\) BaNiS\(_2\). Density-functional-theory-based electronic structure calculations on \(x=0\) BaCoS\(_2\) suggest that these displacements arise as a dynamic first-order Jahn-Teller effect owing to partial occupancy of nominally degenerate Co\(^{2+}\) \(d_{xz}\) and \(d_{yz}\) orbitals, leading to local structural symmetry breaking in the \(xy\)-plane of the Co-rich phases. The Jahn-Teller instability is associated with the opening of a band gap that is further strengthened by electronic correlation. The Jahn-Teller effect is reduced upon increased electron filling as \(x\)→1, indicating that the local structure and band filling cooperatively result in the observed insulator-to-metal transition.

2018
-
 Optimization and Validation of Efficient Models for Predicting Polythiophene Self-AssemblyEvan D. Miller , Matthew L. Jones , Michael M. Henry , Paul Chery , Kyle Miller , and Eric JankowskiPolymers, Dec 2018
Optimization and Validation of Efficient Models for Predicting Polythiophene Self-AssemblyEvan D. Miller , Matthew L. Jones , Michael M. Henry , Paul Chery , Kyle Miller , and Eric JankowskiPolymers, Dec 2018We develop an optimized force-field for poly(3-hexylthiophene) (P3HT) and demonstrate its utility for predicting thermodynamic self-assembly. In particular, we consider short oligomer chains, model electrostatics and solvent implicitly, and coarsely model solvent evaporation. We quantify the performance of our model to determine what the optimal system sizes are for exploring self-assembly at combinations of state variables. We perform molecular dynamics simulations to predict the self-assembly of P3HT at ∼350 combinations of temperature and solvent quality. Our structural calculations predict that the highest degrees of order are obtained with good solvents just below the melting temperature. We find our model produces the most accurate structural predictions to date, as measured by agreement with grazing incident X-ray scattering experiments.
